







![[众诚云网科技]](/uploads/allimg/20190305/c4b08346cbe8b0efae6b132139c2d72a.png)
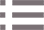
新闻中心 
学一点空间转录组数据分析方法(空间转录组技术方法的比较)
 2023-11-04
2023-11-04 浏览次数:次
浏览次数:次 返回列表
返回列表|撰文:莫北
空间转录组是从组织切片层面上解析RNA-seq数据的新技术,而数据的分析方法却与常规的单细胞转录组有很多相似的地方。在之前的微信文章如《单细胞分组数据分析学习笔记分享》、《单细胞转录组学习笔记之Seurat 3.0(一)》已介绍过如何使用Seurat分析单细胞转录组数据,今天再为大家分享一下Seurat分析空间转录组数据的方法。
本文的范例数据下载链接:
https://support.10xgenomics.com/spatial-gene-expression/datasets本次用到的范例数据为以下两个:Mouse Brain Serial Section 1 (Sagittal-Anterior)Mouse Brain Serial Section 1 (Sagittal-Posterior)
1.数据准备
#载入所需的R包; library(Seurat) library(ggplot2) library(patchwork) library(dplyr) library(hdf5r)数据的读入,可以使用Read10X_h5()或Read10X()读入表达矩阵后创建Seurat对象,方法同常规的单细胞转录组;然后,使用Read10X_Image()函数读入空间图像信息文件,整合到Seurat对象上。比较快速的方式是将表达量数据和图像信息文件置于同一文件夹,用Load10X_Spatial()函数一次性读入,一步到位。
#这里选择“一步到位”的方法: data_dir <-"C:/Users/MHY/Desktop/brain1/" #查看目录中的文件; list.files(data_dir) #[1] "spatial" #[2] "V1_Mouse_Brain_Sagittal_Anterior_filtered_feature_bc_matrix.h5" file_name <-"V1_Mouse_Brain_Sagittal_Anterior_filtered_feature_bc_matrix.h5"注意:路径中不要出现中文!!!如果filename参数不指定,表达量文件的文件名只能是filtered_feature_bc_matrix.h5!
#读入数据并创建Seurat对象; brain <- Load10X_Spatial(data.dir = data_dir, filename = file_name,slice ="anterior1") brain@project.name <-"anterior1" Idents(brain) <-"anterior1" brain$orig.ident <-"anterior1"2.数据的预处理
类似常规单细胞转录组,可用小提琴图展示mRNA分子数和基因数。
p1 <- VlnPlot(brain, features ="nCount_Spatial", pt.size = 0,cols ="tomato") + NoLegend() p2 <- SpatialFeaturePlot(brain, features ="nCount_Spatial") + theme(legend.position ="right") p1 | p2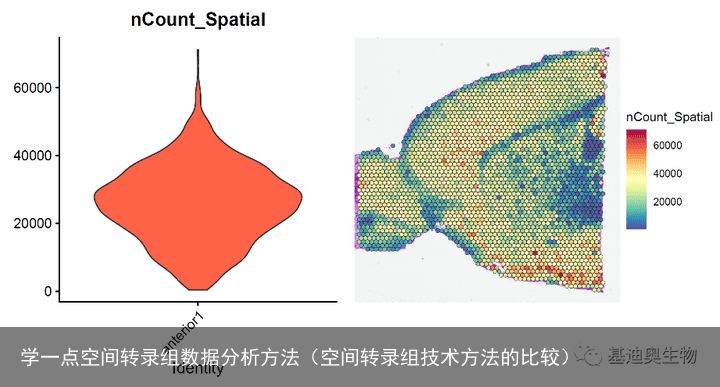 p3 <- VlnPlot(brain, features ="nFeature_Spatial",
pt.size = 0,cols ="tomato") + NoLegend()
p4 <- FeatureScatter(brain, feature1 ="nCount_Spatial",
feature2 ="nFeature_Spatial")+ NoLegend()
p3 | p4
p3 <- VlnPlot(brain, features ="nFeature_Spatial",
pt.size = 0,cols ="tomato") + NoLegend()
p4 <- FeatureScatter(brain, feature1 ="nCount_Spatial",
feature2 ="nFeature_Spatial")+ NoLegend()
p3 | p4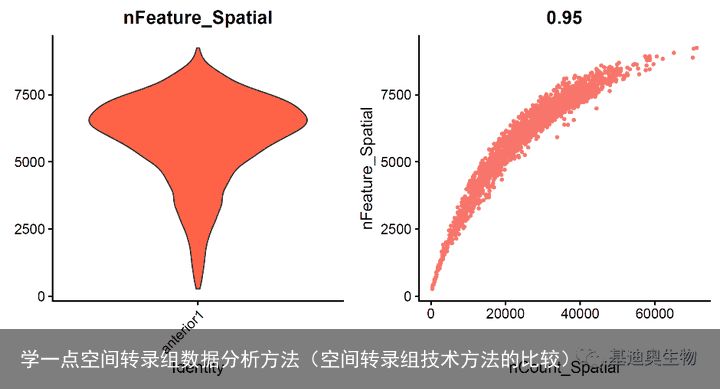
我们发现不同spot的mRNA分子数差异很大,特别是当组织中的细胞密度存在差异时,因此需要对数据进行标准化。由于细胞组织存在异质性,不同组织区域的细胞密度可能不同;因此如果采用常规单细胞转录组数据的LogNormalize标准化方法可能存在问题;这里Seurat作者推荐sctransform方法。
#用SCTransform()对数据进行标准化, 同时检测高变基因, 输出结果储存在 SCT assay中; #耗时约:2min brain <- SCTransform(brain, assay = "Spatial", verbose = FALSE) #提取表达量变变化最高的10个基因; top10 <- head(VariableFeatures(brain),10) top10 #[1] "Ttr" "Plp1" "Hbb-bs" "Mbp" "Hba-a1" "Ptgds" "Penk" "Hba-a2" #[9] "S100a5" "Ppp1r1b" #类似常规转录组,我们也可以展示高变基因; p5 <- VariableFeaturePlot(brain,cols = c("gray60", "red"))+NoLegend() p6 <- LabelPoints(plot = p5,points = top10, repel = TRUE,xnudge=0,ynudge=0) p6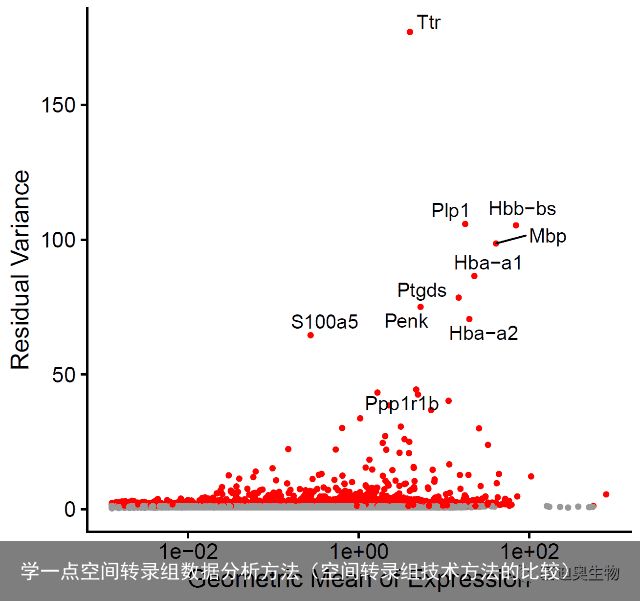
根据空间数据新特性,Seurat加入了新的函数用于展示空间转录组数据,主要方法是将每个spot的表达数据(推测为标准化后的表达量)叠加在组织显微照片上。例如,在本文的小鼠大脑范例数据中,Hpca基因是海马的marker,而Ttr基因是脉络丛的marker。
#展示特定基因的表达水平; SpatialFeaturePlot(brain, features = c("Hpca", "Ttr"))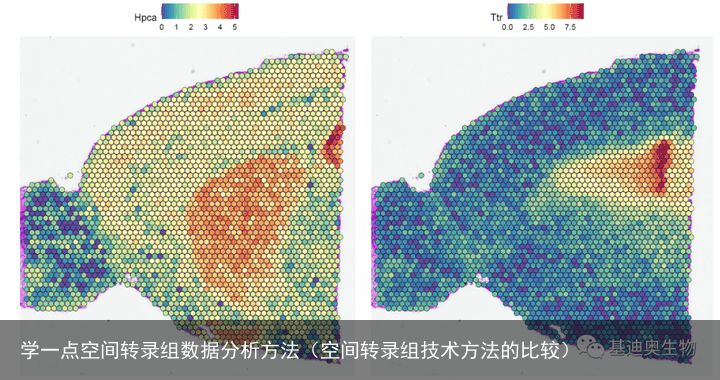 #也可以对“点”进行个性化设置:点的大小和透明度设置;
p7 <- SpatialFeaturePlot(brain, features ="Ttr", pt.size.factor = 1.5,stroke = 0.0)
p8 <- SpatialFeaturePlot(brain, features ="Ttr", alpha = c(0.4, 1))
p7 + p8
#也可以对“点”进行个性化设置:点的大小和透明度设置;
p7 <- SpatialFeaturePlot(brain, features ="Ttr", pt.size.factor = 1.5,stroke = 0.0)
p8 <- SpatialFeaturePlot(brain, features ="Ttr", alpha = c(0.4, 1))
p7 + p8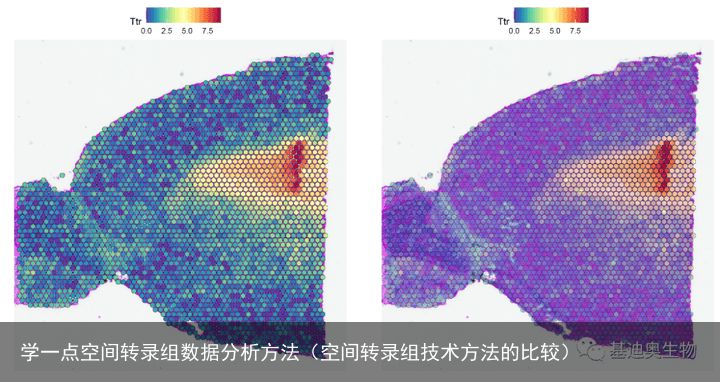
tips:
pt.size.factor:设置spots的大小,默认为1.6;
alpha:设置spots的最小和最大透明度值,默认alpha = c(1, 1),表达量越低越透明;stroke:设置spots的描边粗细,默认stroke = 0.25
3.常规降维聚类流程分析
#接下来是常规单细胞转录组分析流程:PCA降维、UMAP聚类; brain <- RunPCA(brain, assay = "SCT", verbose = FALSE) brain <- FindNeighbors(brain, reduction = "pca", dims = 1:30) brain <- FindClusters(brain, verbose = FALSE) brain <- RunUMAP(brain, reduction = "pca", dims = 1:30) #绘制UMAP分群图; p9 <- DimPlot(brain, reduction = "umap", label = TRUE) p9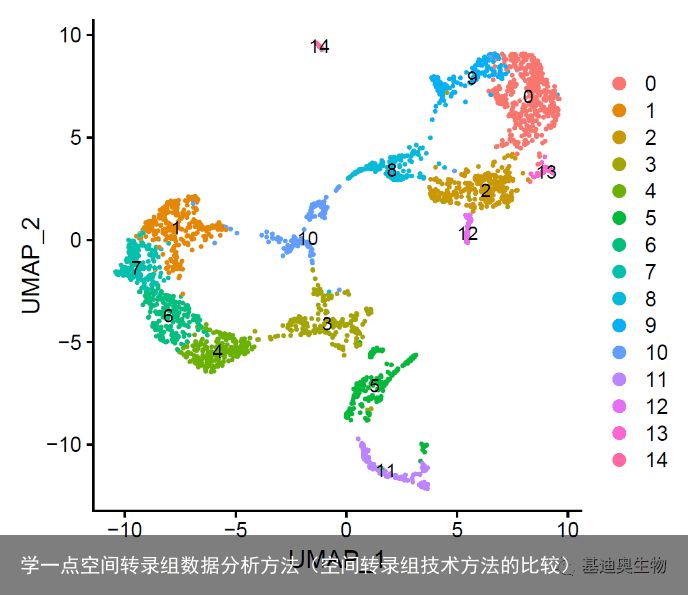 #在切片图像上映射分群信息;
p10 <- SpatialDimPlot(brain, label = TRUE, label.size = 3)
p10
#在切片图像上映射分群信息;
p10 <- SpatialDimPlot(brain, label = TRUE, label.size = 3)
p10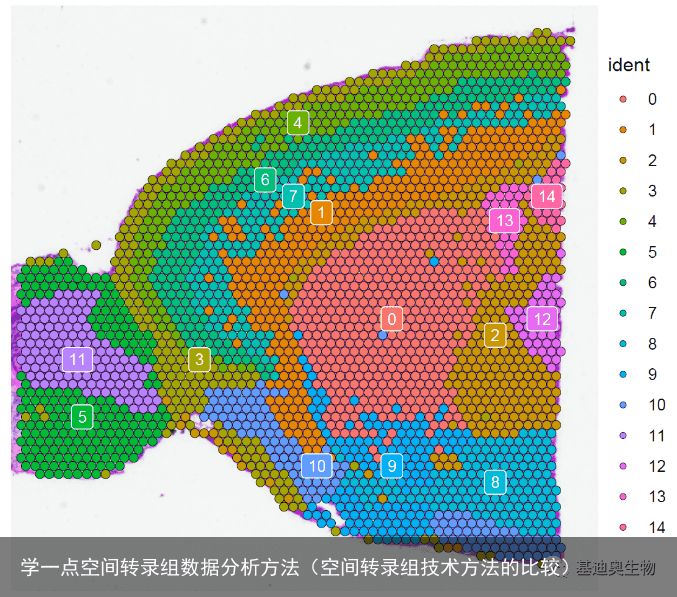 #使用cells.highlight参数单独展示单个cluster的映射情况,这里仅展示1~4个cluster;
cluster <- CellsByIdentities(object = brain, idents = c(1, 2, 3, 4))
SpatialDimPlot(brain, cells.highlight = cluster ,
facet.highlight = TRUE,
cols.highlight = c("green","grey70"),
ncol = 2)
#使用cells.highlight参数单独展示单个cluster的映射情况,这里仅展示1~4个cluster;
cluster <- CellsByIdentities(object = brain, idents = c(1, 2, 3, 4))
SpatialDimPlot(brain, cells.highlight = cluster ,
facet.highlight = TRUE,
cols.highlight = c("green","grey70"),
ncol = 2)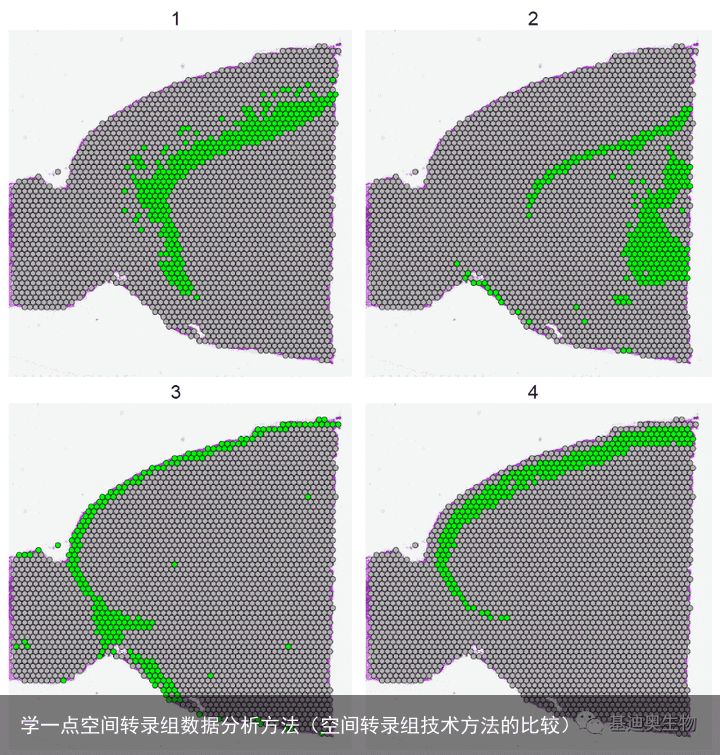
4.差异分析
#对5、6两个cluster进行差异分析; de_markers <- FindMarkers(brain, ident.1 = 5, ident.2 = 6) head(de_markers) #绘制前3个差异基因的表达映射图;
SpatialFeaturePlot(object = brain,
features = rownames(de_markers)[1:3],
alpha = c(0.1, 1), ncol = 3)
#绘制前3个差异基因的表达映射图;
SpatialFeaturePlot(object = brain,
features = rownames(de_markers)[1:3],
alpha = c(0.1, 1), ncol = 3) #无监督查找marker基因,提取前面生成的3000个高变基因;
genes <- VariableFeatures(brain)
#从前1000个中查找差异基因;
#若是1000个,耗时约30min(这里演示只用前100个);
brain <- FindSpatiallyVariableFeatures(brain, assay = "SCT",
features = genes[1:100],
selection.method = "markvariogram")
DEGs <- SpatiallyVariableFeatures(brain, selection.method = "markvariogram")
top.features <- head(DEGs, 6)
#对差异基因进行可视化;
SpatialFeaturePlot(brain, features = top.features, ncol = 2, alpha = c(0.1, 1))
#无监督查找marker基因,提取前面生成的3000个高变基因;
genes <- VariableFeatures(brain)
#从前1000个中查找差异基因;
#若是1000个,耗时约30min(这里演示只用前100个);
brain <- FindSpatiallyVariableFeatures(brain, assay = "SCT",
features = genes[1:100],
selection.method = "markvariogram")
DEGs <- SpatiallyVariableFeatures(brain, selection.method = "markvariogram")
top.features <- head(DEGs, 6)
#对差异基因进行可视化;
SpatialFeaturePlot(brain, features = top.features, ncol = 2, alpha = c(0.1, 1))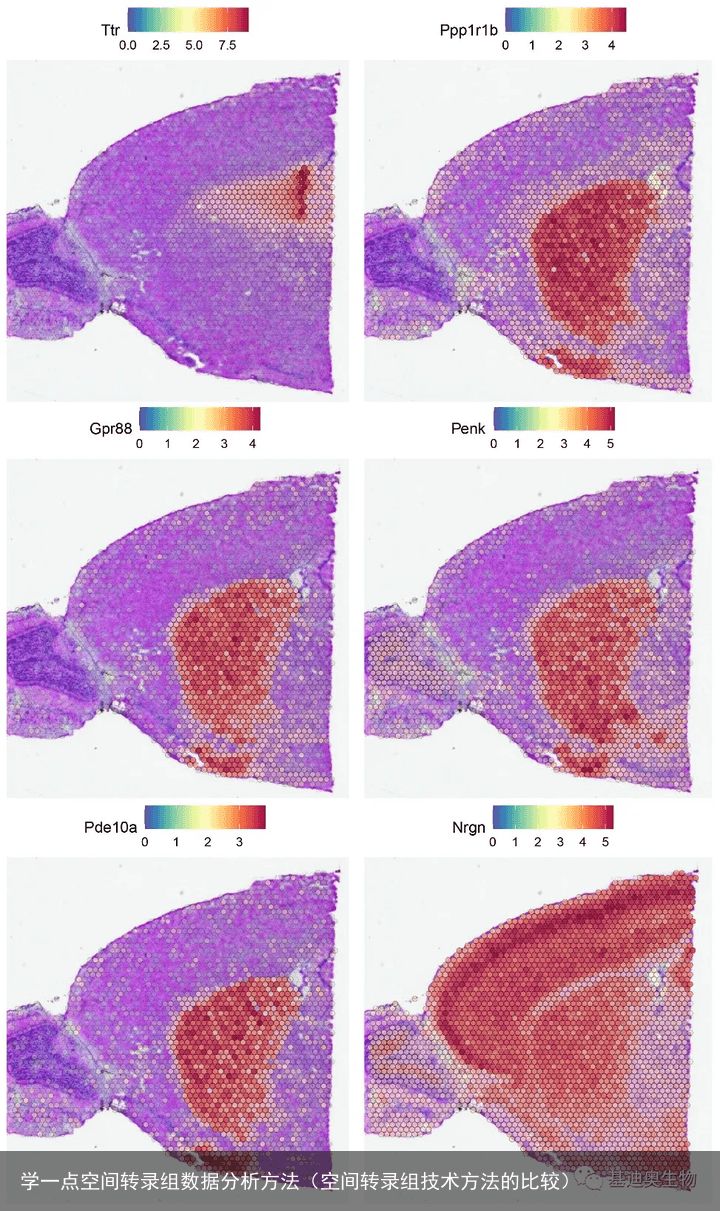
5.提取子亚群
#这里,我们先粗略提取额叶皮层的spots数据,然后根据坐标逐步“精修”; cortex <- subset(brain, idents = c(1, 2, 3, 4, 6, 7)) p_img <- SpatialDimPlot(cortex, crop = TRUE, label = TRUE, label.size = 3) p_img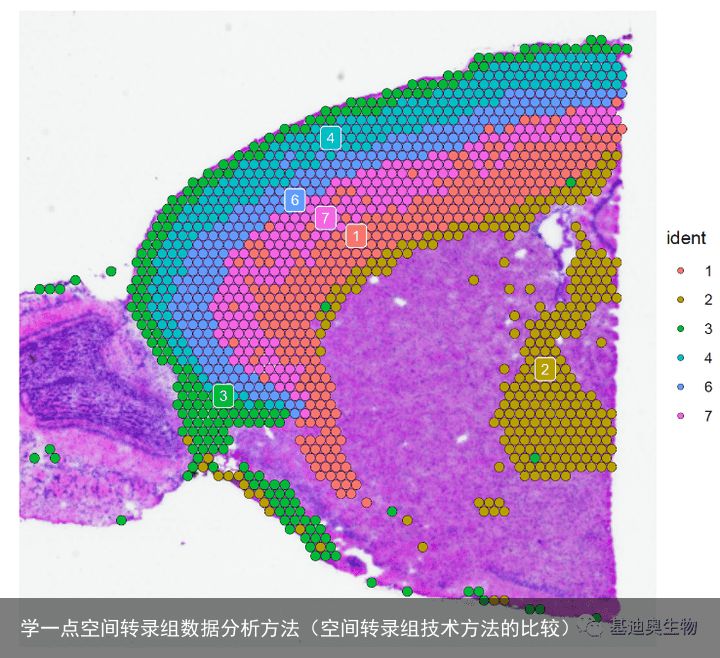 #获取切片图像坐标和Key:
Key(object =cortex@images$anterior1)
img<- GetTissueCoordinates(cortex)
head(img)
#增加分群信息;
ident <- Idents(cortex)
imgId <- data.frame(img,ident)
head(imgId)
#获取切片图像坐标和Key:
Key(object =cortex@images$anterior1)
img<- GetTissueCoordinates(cortex)
head(img)
#增加分群信息;
ident <- Idents(cortex)
imgId <- data.frame(img,ident)
head(imgId)
显微照片的像素坐标原点在左上角,而图表的原点在左下角,因此需要做垂直翻转。
#spots坐标信息的探索; p_plot <- ggplot(img, aes(x=imagecol, y=600-imagerow,color = ident)) + geom_point(size = 0.6) p_img+p_plot#根据切片图像坐标(前缀需和Key保持一致)进一步去除多余的spots数据; cortex <- subset(cortex, anterior1_imagerow > 400 | anterior1_imagecol < 150, invert = TRUE) SpatialDimPlot(cortex, crop = TRUE, label = TRUE, label.size = 3) cortex <- subset(cortex, anterior1_imagerow > 275 & anterior1_imagecol > 370, invert = TRUE) SpatialDimPlot(cortex, crop = TRUE, label = TRUE, label.size = 3) cortex <- subset(cortex, anterior1_imagerow > 250 & anterior1_imagecol > 440, invert = TRUE) SpatialDimPlot(cortex, crop = TRUE, label = TRUE, label.size = 3)#对切片特定区域的数据进行局部和整体可视化; p11 <- SpatialDimPlot(cortex, crop = TRUE, label = TRUE, label.size = 3) p12 <- SpatialDimPlot(cortex, crop = FALSE, label = TRUE, pt.size.factor = 1, label.size = 3) p12 + p116.多切片数据合并
接下来读入另一个切片的数据,并创建对象,方法同上。
data_dir <- "C:/Users/MHY/Desktop/brain2/" #查看目录中的文件; list.files(data_dir) file_name <- "V1_Mouse_Brain_Sagittal_Posterior_filtered_feature_bc_matrix.h5" #读入数据并创建Seurat对象; brain2 <- Load10X_Spatial(data.dir = data_dir, filename = file_name,slice="Posterior1") brain2@project.name <- "Posterior1" Idents(brain2) <- "Posterior1" brain2$orig.ident <- "Posterior1" #读入数据并对表达量数据标准化; brain2 <- SCTransform(brain2, assay = "Spatial", verbose = FALSE) #合并两个对象; brain.merge <- merge(brain, brain2) save(brain.merge,file = "brain.merge_sct.Rdata")Tips:对于本案例,个人电脑执行此操作需确保内存足够(至少8G)并关掉其他消耗资源的软件(如腾讯会议),避免电脑卡死。
#合并高变基因集,执行常规的PCA、分群聚类流程; DefaultAssay(brain.merge) <- "SCT" VariableFeatures(brain.merge) <- c(VariableFeatures(brain), VariableFeatures(brain2)) brain.merge <- RunPCA(brain.merge, verbose = FALSE) brain.merge <- FindNeighbors(brain.merge, dims = 1:30) brain.merge <- FindClusters(brain.merge, verbose = FALSE) brain.merge <- RunUMAP(brain.merge, dims = 1:30) #对合并后的分群数据进行可视化; DimPlot(brain.merge, reduction = "umap", group.by = c("ident", "orig.ident"))#比较两个切片的分群情况; #默认使用两个slice的名称分组; SpatialDimPlot(brain.merge) #如果创建对象时没有对slice重命名,也可使用ident进行分组,绘图效果一样; SpatialDimPlot(brain.merge,group.by = c("ident"))#比较两个切片的特定基因表达水平; SpatialFeaturePlot(brain.merge, features = c("Hpca","Plp1"))好啦,以上就是全部的分享内容啦!想学习更多单细胞数据分析技巧,欢迎报名基迪奥生物的单细胞培训班~
|培训时间:2020年12月14日-12月18日
|报名费:1800元
优惠:
团购价:1500元,2人或2人以上报名,可享受团购价
|报名方式:
发送姓名、学校、电话到邮箱contact@genedenovo.com,主题注明“单细胞培训班”报名截止时间:2020年12月8日
客服电话:020-39341079或18054271626 邓小姐
新一期的单细胞转录组培训班课程安排如下:
